Building a Scalable Reference Framework for a High-Quality Bat Genome Assembly
Overview
Excelra partnered with a biotech innovator to develop a scalable, automated pipeline for high-quality bat genome assembly. Utilizing PacBio HiFi and Hi-C sequencing data, the pipeline achieved >98% genome completeness and >99.9% base-level accuracy. It integrated long-read and Hi-C data for chromosome-scale assemblies and was modularly designed to support viral genome assembly as well. The solution reduced manual effort by 75% and accelerated early discovery and therapeutic target identification. With validation-ready outputs and cross-platform compatibility, Excelra’s framework empowered the client to scale genomic research efficiently, enhancing their competitive edge in bat immunogenomics.

Our client
A biotech innovator at the forefront of bat genome research, studying evolutionary adaptations. By leveraging bat genomics, the client aimed to develop therapeutic strategies, uncovering unique biological traits and immune pathways. The client, in early discovery stages, required technical acceleration through bioinformatics automation. Excelra was tasked with providing a framework to automate the assembly of PacBio HiFi sequencing reads plus Hi-C data to construct reliable bat genome assemblies.

Client’s challenge
An early-stage biotech company had generated large datasets from bat genome sequencing. Bats, with over 1,400 species, show significant genome divergence, complicating comparative genomics and reference-guided assembly. High-quality de novo genome assembly using long-read sequencing technologies was critical to accurately capture shared and species-specific genomic features.
Related case study: Scalable Bat Genome Framework

Client’s goals
Wanted to build an efficient high-quality workflow for automating the assembly process of reference genome assemblies from scratch. They required a robust, automated pipeline to:
- Assemble PacBio HiFi reads
- Integrate Hi-C data for scaffolding
- Enable consistent and scalable genome assembly workflows
- Extend the framework to support virus genome assemblies from Illumina RNA-Seq data
Our approach
Excelra designed and deployed an end-to-end genome assembly pipeline tailored to high-accuracy PacBio HiFi sequencing data and Hi-C scaffolding, also supporting viral genome assembly from short-read data. The engagement aimed to standardize the de novo assembly pipeline for bat genomes using PacBio, 10X, Bionano, and Hi-C technologies.
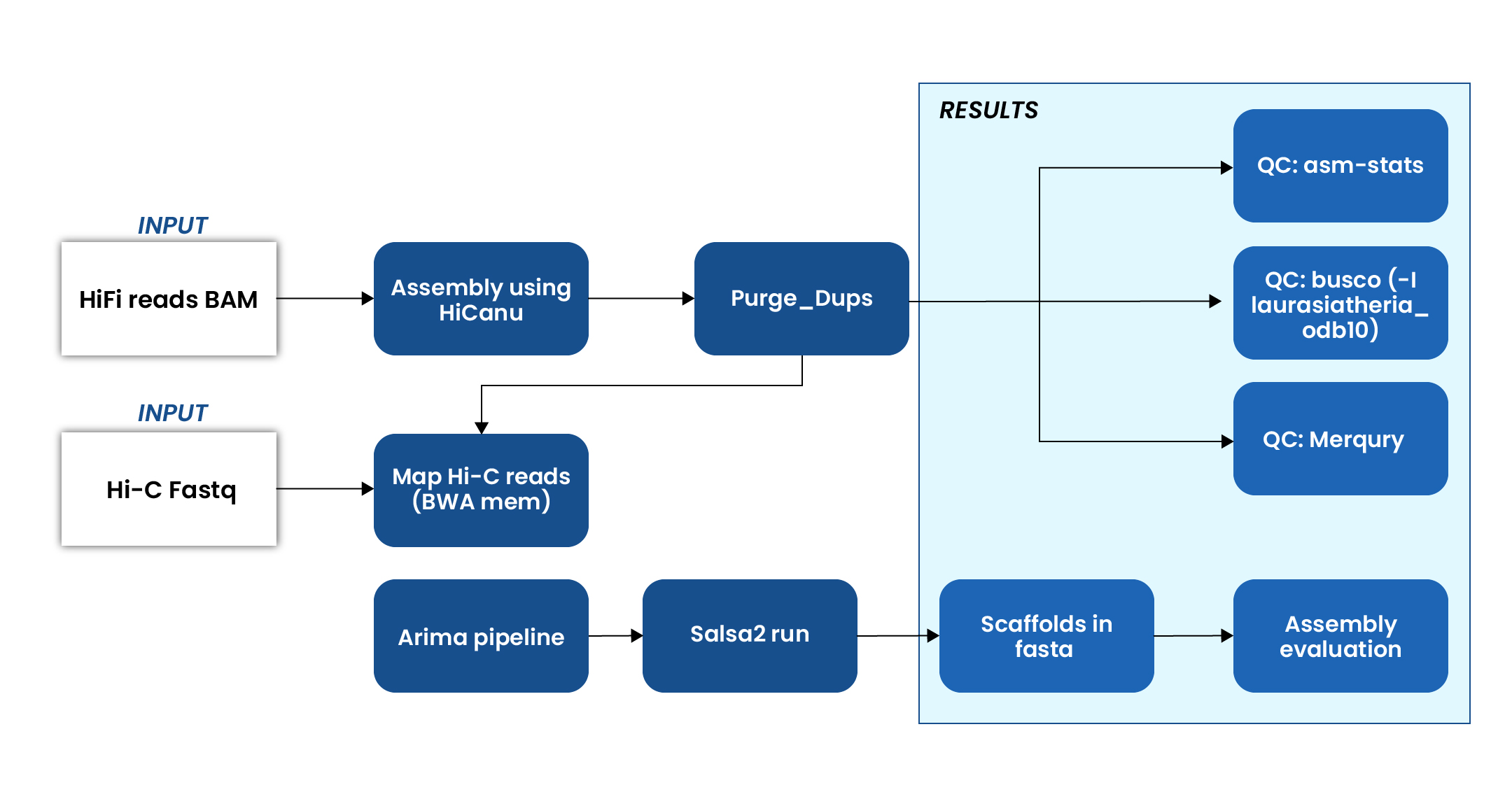
Genome assembly workflow
Input data
- PacBio HiFi BAM files for high-accuracy long-read sequencing.
- Hi-C fastq files for capturing chromatin interaction patterns.
HiFi assembly:
- Performed initial genome assembly using a tool, optimized for PacBio HiFi reads.
- Cleaned the assembly to remove haplotypic duplications and improve contiguity.
Hi-C integration
- Aligned Hi-C reads to the draft assembly using a tool for accurate mapping.
- Processed the mapped Hi-C data to prepare scaffolding input.
- Scaffolded the assembly using a tool, which utilizes Hi-C contact data to order and orient contigs into chromosome-scale scaffolds.
Assembly quality assessment
Conducted a comprehensive evaluation of the genome assembly using multiple tools to ensure accuracy, completeness, and structural integrity:
- Generated key assembly statistics such as N50, total assembly length, and number of contigs/scaffolds, providing a snapshot of assembly contiguity.
- BUSCO (Benchmarking Universal Single-Copy Orthologs) analysis was performed using the laurasiatheria_odb10 lineage dataset to assess the completeness of the assembly in terms of conserved gene content.
- Estimated base-level accuracy and completeness using a k-mer-based approach, comparing the assembly to raw sequencing reads for validation.
Final outputs
- A scaffolded genome in FASTA format, representing the assembled and Hi-C–scaffolded genome sequence at chromosome-level resolution.
- K-mer multiplicity plots, used to visualize and validate the consistency between the raw read data and the final assembly, helping to identify potential errors, duplications, or missing regions.
Related whitepaper: Omics Data: A Biomedical Asset Driving the Future of Drug Discovery
Our solution
Delivered a modular, automated pipeline for de novo assembly of bat genomes using PacBio HiFi reads and Illumina Hi-C reads , enabling:
- High-quality, reproducible reference genomes
- Smooth integration of long-read and Hi-C data
- Validation-ready outputs for downstream research. The assembly metrics, reads k-mer validation, and orthologous genes validation show that we were able to assemble a correct genome from HiFi and Hi-C reads.
-
Key performance outcomes
>98% assembly completeness, confirmed the presence of most conserved genes in the assembled genome.
>99.9% base-level accuracy, ensured high fidelity of the assembly with minimal sequencing or structural errors.
Reduced manual effort and turnaround time by 75%, enabling rapid generation of reference-quality genomes for multiple bat species.
Cross-platform data integration, reduced integration complexity and data processing costs.
Modular design for reusability, reproducible and scalable genome assembly workflows for future species or projects, eliminating the need to re-engineer pipelines.

Strategic business impacts
- Accelerated early discovery pipeline: High-quality assemblies enabled faster comparative and functional genomics research.
- Enhanced therapeutic target discovery: Accurate genomes unlocked unique biological traits for novel therapeutic target identification.
- Future-ready infrastructure: Modular pipeline supports future genome assemblies for bats and related pathogens.
- Improved data confidence for downstream analysis: Validation-ready outputs boosted confidence in downstream analyses with minimal additional processing.
- Strengthened competitive advantage: Early access to high-fidelity genomes positioned the client ahead in bat immunogenomics research.
Conclusion
Excelra delivered a scalable, automated genome assembly pipeline, enabling a biotech client to generate high-quality, chromosome-scale bat genome assemblies with >98% completeness and >99.9% accuracy. The modular, cross-platform design allowed rapid scaling of future genome projects without re-engineering. This solution accelerated early discovery, enhanced therapeutic target identification, and provided validation-ready outputs, strengthening the client’s competitive position in bat immunogenomics research.
Related internal links: Pipeline for Immune Repertoire Data Analysis, Bioinformatics for Drug Discovery, Data-Driven Drug Discovery.
