Development of Optimized Workflow for Viral Genome Reconstruction and Taxonomic Classification from RNA-Seq Data
Overview
We partnered with a pioneering biotech company focused on bat genomes to develop an automated, scalable workflow for viral genome assembly and taxonomic classification from Illumina RNA-Seq data. By integrating advanced bioinformatics workflow managers and a dual-path assembly strategy, the pipeline achieved over 95% sequence identity and 98% classification accuracy. This solution not only accelerated virus detection and genome reconstruction but also enhanced zoonotic surveillance, supporting early identification of potential spillover threats. The project underscores Excelra’s commitment to enabling faster, data-driven decisions in virology, public health, and vaccine development, in line with AI-driven data analysis for precision medicine.

Our client
Our client is a biotech innovator at the forefront of bat genome research, studying evolutionary adaptations. The client needed Excelra’s expertise to assemble viral genomes embedded within host transcriptomic RNA-Seq data and perform accurate taxonomic classification to generate high-quality reference sequences.

Client’s challenge
The early-stage biotech company faced challenges in automating the assembly of Illumina RNA-Seq reads from host species to construct reliable virus genome assemblies and perform taxonomic classification. Species like bats and swine act as natural reservoirs for diverse viruses, some with spillover potential, as highlighted during pandemics like COVID-19. RNA-Seq data from these hosts is a crucial resource for early virus identification and genomic data management.

Client’s goals
By analyzing RNA-Seq data across species, researchers can gain insights into which viruses may pose a risk of spillover, helping pharmaceutical companies and public health organizations prepare for emerging threats.
The client sought to feed this RNA-Seq data into a virus genome assembly pipeline to create an automated, scalable workflow. The resulting viral genome assemblies would not only support their current research studying taxonomic classification but also serve as high-quality reference sequences for future studies. The client’s goal was to study virus integration in bats constructing reliable virus genome assemblies from host-derived RNA-Seq data.
Client’s goals
By analyzing RNA-Seq data across species, researchers can gain insights into which viruses may pose a risk of spillover, helping pharmaceutical companies and public health organizations prepare for emerging threats.
The client sought to feed this RNA-Seq data into a virus genome assembly pipeline to create an automated, scalable workflow. The resulting viral genome assemblies would not only support their current research studying taxonomic classification but also serve as high-quality reference sequences for future studies. The client’s goal was to study virus integration in bats constructing reliable virus genome assemblies from host-derived RNA-Seq data.
Our approach
Excelra designed a robust and automated pipeline to process Illumina RNA-Seq reads and assemble viral genomes. The workflow leveraged multiple bioinformatics tools and databases to streamline assembly and taxonomic analysis:
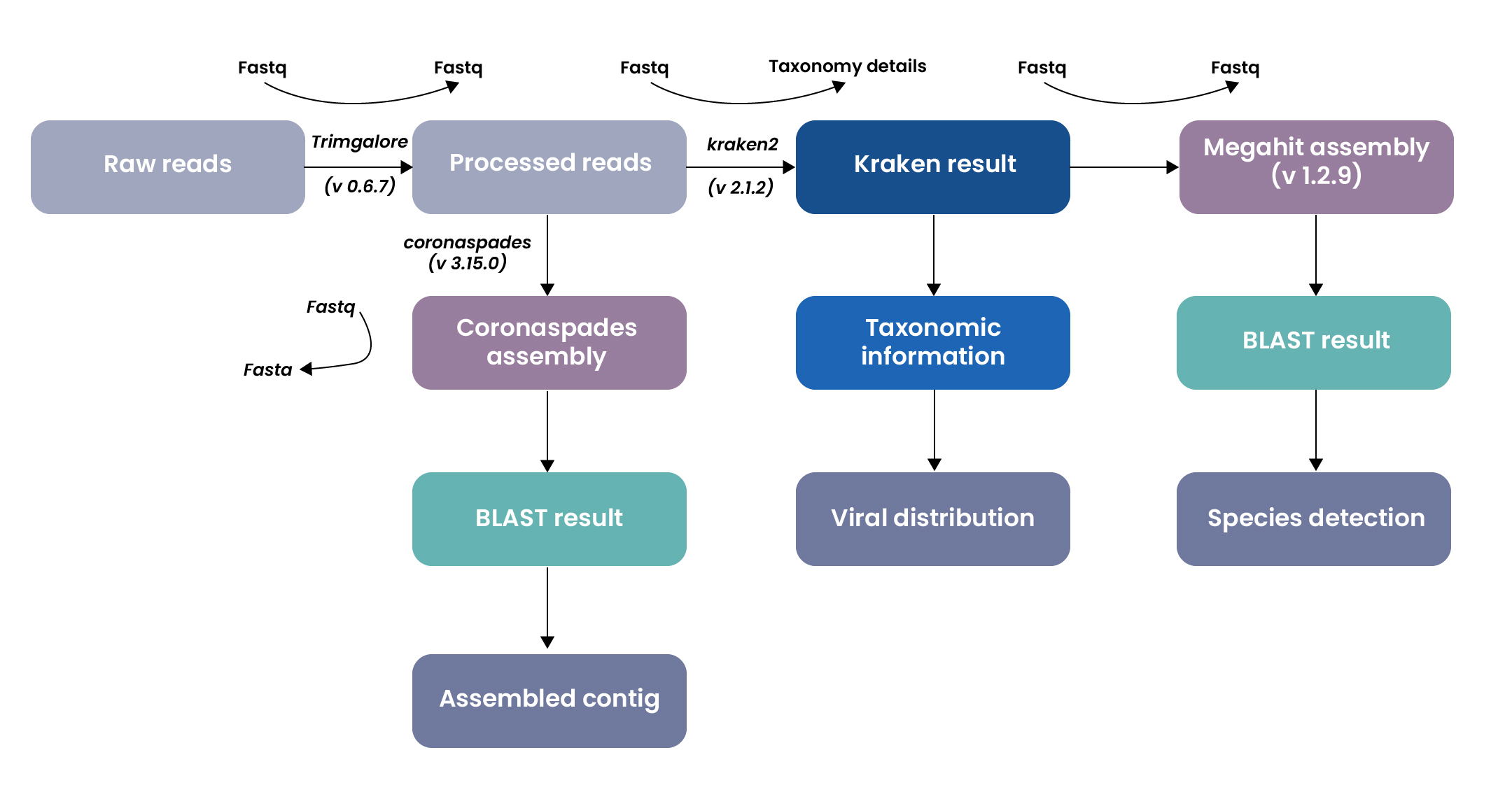
Pre-processing
- Raw sequencing reads were subjected to quality trimming.
- This step removes low-quality bases and adapter contamination, ensuring that only high-quality, clean reads are used for downstream analysis.
Assembly
The cleaned reads were processed through two complementary assembly strategies to maximize the detection and reconstruction of viral genomes:
- A general-purpose metagenome assembler was employed, ideal for generating a broad and comprehensive assembly of all organisms present in the sample.
- A virus-focused assembler optimized for RNA viruses, was specifically used to reconstruct viral genomes with higher sensitivity, particularly those related to coronaviruses and similar viral pathogens.
Annotation & taxonomic classification
- A fast and accurate taxonomic classifier, was used to identify and classify the microbial and viral components present in the sample, providing taxonomic labels to the assembled sequences.
- BLAST analysis was then carried out on the assembled contigs from above processes to: Confirm the taxonomic identities and validate the presence of known or novel viral species through sequence similarity against reference databases.
Quality analysis & interpretation:
The final assemblies were evaluated for:
- Taxonomic composition—to determine the diversity and abundance of organisms present.
- Viral genome distribution—to assess the breadth of viral detection across different taxa.
- Contig assembly quality—to ensure the assembled viral genomes are contiguous, complete, and biologically meaningful.
These evaluations helped to ensure confidence in the detection, classification, and downstream interpretation of viral and microbial sequences within the sample.
Our solution
Excelra successfully assembled virus genomes with high percentage identity to references using our workflow with Illumina RNA-Seq reads. This robust workflow enables comprehensive detection and reconstruction of viral sequences from complex sequencing datasets, ensuring both accuracy and biological relevance. The key components of the pipeline include:
High identity assemblies
Successfully reconstructed viral genomes with high sequence identity to known references using Illumina RNA-Seq data.
Comprehensive detection & reconstruction
Workflow ensures accurate identification and biologically relevant reconstruction of viral sequences from complex datasets.
Quality trimming & pre-processing
- Applied stringent quality control.
- Removed low-quality bases and adapter sequences using standard tools.
Dual-path assembly strategy
- General-purpose assembler (e.g., MEGAHIT): Captured broad sequence diversity.
- Virus-specific assembler (e.g., coronaSPAdes): Optimized for sensitive reconstruction of RNA viral genomes.
Taxonomic classification via Kraken2
- High-performance k-mer-based classification of contigs.
- Enabled detection of both known and novel viral species.
Downstream analysis included
- BLAST validation for confirming viral identities.
- Distribution mapping to assess viral abundance and prevalence.
- Species-level detection to resolve closely related strains.
Final output
Delivered high-quality, assembled viral contigs with validated annotations and taxonomic labels—supporting virome studies, epidemiology, and functional research.
Key performance outcomes
>95% sequence identity achieved to known reference genomes, ensuring high-confidence detection of both known and novel viruses.
>98% taxonomic classification accuracy in identifying viral species and strains.
>3x improvement in assembly turnaround time compared to manual or semi-automated methods.
Early discovery of potential zoonotic threats, providing a 6–12 months lead time for risk mitigation compared to traditional surveillance.
Up to 70% reduction in manual curation effort, freeing up bioinformatics experts for higher-value tasks.

Strategic business impacts
- Enhanced pandemic preparedness through high-throughput zoonotic surveillance
- Faster vaccine and antiviral development powered by high-quality reference genomes
- Creation of proprietary genomic assets for R&D, licensing, and partnership opportunities
- Improved regulatory support via quality-controlled genome assemblies for submissions and surveillance
Conclusion
Excelra delivered a customized, automated bioinformatics pipeline that enabled accurate and efficient assembly of viral genomes from host-derived RNA-Seq data. The solution significantly improved processing speed, reproducibility, and scalability, while achieving >95% sequence identity to reference genomes and >98% taxonomic classification accuracy. Designed for modular integration, the pipeline is capable of processing >10,000 datasets annually with up to 70% reduction in manual curation. This empowers large-scale viral genomics efforts—particularly in zoonotic reservoirs like bats—supporting early detection of emerging threats with a 6–12 months lead time. Additional value delivered to the client includes accelerated vaccine and antiviral target discovery, proprietary data asset creation, and robust support for regulatory submissions.
