Poor drug solubility is one of the main obstacles in drug discovery and development and is strongly related to the choice of target explored. (Bergstrom et al., 2016). Solubility is critical for absorption and acceptable solubility in the intestinal fluid is required to achieve sufficiently high drug blood concentrations to obtain a therapeutic effect when systemic effects are warranted. The solubility of a compound affects its absorption, distribution, metabolism, excretion and toxicity (ADMET) profile. Only drug candidates whose ADMET properties are of sufficient quality can be further developed.
SAR databases
Machine learning methods are reshaping research on the properties of molecules. The better the datasets that are used to train and test these methods, the more robust the results are expected to be. In this white paper, we compare two such SAR databases: GOSTAR®, which is the largest Structure Activity Relationship (SAR) database for drug discovery, and MoleculeNet, which comprises multiple public datasets, establishes metrics for evaluation and offers open-source implementations of multiple previously proposed algorithms for evaluating compounds using machine learning.
The MoleculeNet collection includes over 700,000 compounds tested on a range of different properties. The benchmark also tests the performances of various machine learning models with different featurizations on the datasets and results reported in AUC-ROC, AUC-PRC, RMSE and MAE scores.
GOSTAR® (Global Online Structure Activity Relationship) provides a 360⁰ view of over 8 million small molecule discovery compounds and close to 50,000 preclinical/ clinical candidates, and approved drugs. Content in GOSTAR® is meticulously curated manually from various published data sources with information on chemical structures and their biological properties that includes binding, in-vitro, in-vivo, ADME, tox, and physicochemical properties.
Datasets (LogD and Solubility)
Our study, focused on solubility, started with over 9,000 molecules from the GOSTAR® dataset, approximately 6,800 of which were unique.
After data cleaning and preprocessing, the total set of valid records contains 21360 for the GOSTAR® data and 4200 for MoleculeNet data.
These are the dataset we used for the benchmark in this work.
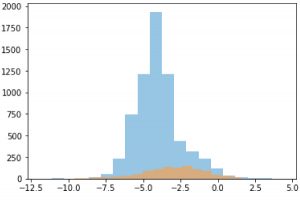
Distribution of solubility data from GOSTAR® and MoleculeNet
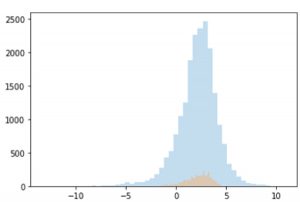
Figure 2. distribution of LogD data from GOSTAR® and MoleculeNet
Methodology
GlamorousAI used its flagship platform, RosalindAI, to build, train and evaluate a zoo of machine learning models that predict solubility and logD. RosalindAI is an end-to-end platform for managing molecular data and developing cutting-edge ML pipelines in a scalable and reproducible manner without the need for coding skills. RosalindAI automatically ingests molecular data and executes pipelines of automated processes and modeling that include data cleaning and preprocessing, featurization, model initiation, hyper-parameter optimization, and model benchmarking.
For the purpose of this study, RosalindAI is used to clean and prepare the data, develop and train the models and report the results. We used random forest for benchmarking as it is the golden standard. For best performance, we used a zoo of models that includes different types of deep learning models trained in a supervised manner on different molecular featurisations (SMILES, Graph, RDKit, Morgan, etc). RosalindAI allows testing of a large number of modeling architecture and hyperparameter optimization strategies to ensure convergence to the best model.
Results and discussion
GlamorousAI Result Set
To assess the impact of GlamorousAI we completed two test phases.
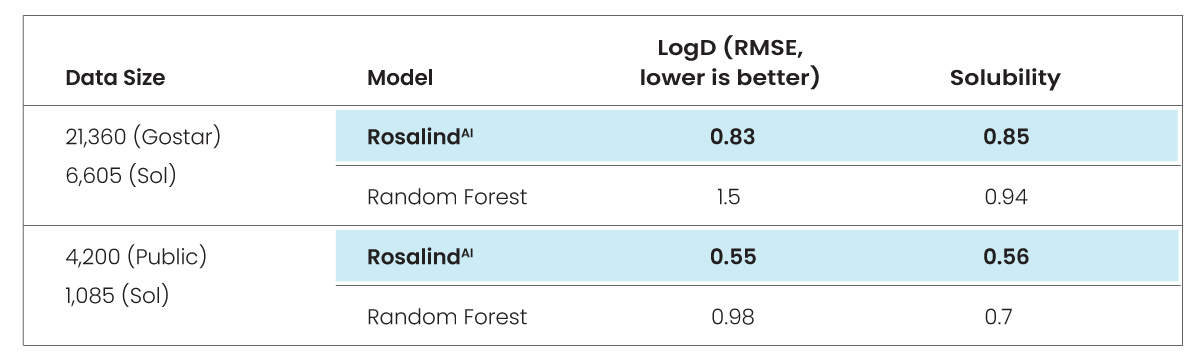
First, we first trained GlamorousAI and Random Forest with GOSTAR® data, then repeated the process with public data and compared the results using root mean square error (RMSE) methodology.
RMSE measures the difference between values predicted by a model and those actually observed. The lower the RMSE, the better the model is at predicting values.
We then assessed the comparative impact of using a large, diverse data set versus a small, concentrated dataset. The test compared 6,605 GOSTAR® molecules against 1,085 from MoleculeNet.
The test concludes that using a large dataset such as GOSTAR®’s, GlamorousAI models are 9.5% better than Random Forest. They are also 14% better than Random Forest when using a smaller public dataset. It is clear, then, that GlamorousAI models are more accurate than Random Forest, regardless of the size of the dataset.
The bottom line following both phases of the test was that GlamorousAI models built on GOSTAR® data were approximately 90x better at predicting solubility than the ones built from public data.
Many off-target liabilities, such as plasma protein binding (especially albumin), hERG, CYP interactions, and transporters, have strong correlations with lipophilicity, and a number of studies have linked high logD to the likelihood of compounds failing in development due to poor ADMET (absorption, distribution, metabolism, excretion and toxicity) characteristics. The
majority of known drugs contain ionizable groups and are likely to be charged at physiological pH. The distribution constant, LogD, is therefore a better descriptor of a molecule’s lipophilicity than logP. LogD is thus pH dependent, so the pH at which the logD was measured must be specified. As the physiological pH of blood serum is 7.4, logD7.4 is of particular interest.
Conclusion
It’s clear from this study that GOSTAR®’s proprietary data set combined with rigorous processes for data cleaning and large scale ML training and development deliver far more robust and actionable results. The larger number of data points, intensive curation, and available trouble-shooting for cleaning up data make this a much better user experience. Side-to-side studies of GlamorousAI application to GOSTAR® and public data for solubility analysis shows that GlamorousAI provides substantially better results with either dataset.
References
- Chen H, Engkvist O, Wang Y et al (2018) The rise of deep learning in drug discovery. Drug Discov Today 23. https://www.sciencedirect.com/science/article/pii/S1359644617303598
- Bhhatarai B, Walters WP, Hop C et al (2019) Opportunities and challenges using artificial intelligence in ADME/Tox. Nat Mater 18. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6594826/
- Tang B, Kramer ST, Fang M et al (2020) A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. J Cheminform 12. https://jcheminf.biomedcentral.
com/articles/10.1186/s13321-020-0414-z - Jiang D, Wu Z, Hsieh C-Y, et al (2021) Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J Cheminform
13. https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00479-8#Sec24 - Wu Z, Ramsundar B, Feinberg E N, et al (2017) MoleculeNet: a benchmark for molecular machine learning. arXiv.org