Artificial intelligence (AI) and machine learning (ML) have the potential to revolutionize the drug discovery and development process. AI/ML simulations are helping researchers identify promising drug candidates and predict how they will behave in the body, reducing the time and cost associated with bringing a new drug to market.
This white paper explores the role of AI and ML in predictive modeling for drug discovery and development, highlighting the benefits and challenges of using these technologies. It discusses the impact data selection has on the predictive accuracy of AI/ML models and the importance of structuring and preparing data so that predictive models can effectively utilize it. Finally, it presents a workflow for selecting and preparing GOSTAR® data to build faster and more accurate AI/ML predictive models.
AI/ML in drug discovery and development
The use of AI/ML models in drug discovery and development has increased substantially in recent years. Drug development is a notoriously slow process, requiring huge investment for slim odds of a return. Any emerging technology that promises to reduce cost, increase speed, and improve the odds of success will obviously be welcomed enthusiastically by the biopharmaceutical industry. That’s undoubtedly been the case with AI/ML modeling, which is now an integral part of the drug discovery and development process. The use of predictive models has surged over the past decade to the point at which it would be inconceivable to commit to a development program without incorporating a significant phase of AI/ML modeling.
The power of AI/ML is utilized across the development program. Readers are directed to other comprehensive reviews on the use of ML in drug discovery and development.[1-4]
But of equal importance is the proliferation of data with which to train models and create algorithms. AI/ML models can quickly ingest clinical data from a wide range of sources and recognize patterns that may be hidden from the human eye. By analyzing these patterns and simulating potential outcomes multiple times, models can make predictions about the efficacy of treatments long before they are trialed on organic subjects.
Improving the accuracy of those predictions becomes one of the primary objectives of biopharmaceutical R&D teams. Greater computational power helps. The more instructions per second (IPS) a processor can execute, the more a prediction can be tested, analyzed, and refined. But instead of idly waiting for processing speeds to multiply, researchers can proactively increase the accuracy of their predictive engines today by improving the quality of the fuel that powers them – the data.
Choosing the best data sets for AI/ML predictive models
The time-saving benefits of ML modeling have been clearly established. Yet building an ML model is itself a laborious process. Creating the algorithm is relatively straightforward, but the inherent inefficiency is the processing and cleansing of data. In fact, approximately 80% of the time building an ML model is spent preparing data and only 20% writing the algorithm. Data processing and cleaning is the bottleneck and limiting factor in the ML approach, underlining the need for large volumes of high-quality, annotated data to optimize the model [Figure 1].[2]
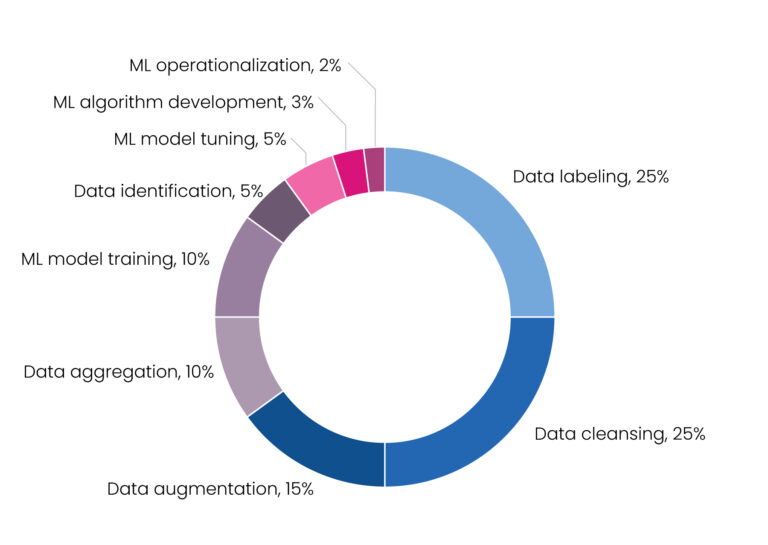
Figure 1. Percentage of time allocated to machine learning projects[5]
Data is the raw material on which AI/ML models are built and operated. But the quality of data is widely variable, and the heterogeneity of data sets – gathered from dozens of global sources at the expense of great resources – can be difficult to standardize and optimize for AI/ML analysis.
Above all, predictive accuracy relies on three key characteristics that databases must provide to facilitate high-performing models: quantity, quality, and coverage.
These three characteristics must be balanced to suit a model’s specific task. Without a sufficiently high volume of data, a model’s prediction is invalidated by its sample size. The qualifying criteria for a valid prediction will change depending on the type of prediction required. At the higher end, for example, an extremely large volume of data would be required for a model built to differentiate between active and inactive compounds across a highly diverse set of chemicals. At the other end of the spectrum, a lower number of data points are required for a refined quantitative model to optimize molecular interactions between a compound and its receptor as measured by x-ray crystallography. Yet, in both cases, data coverage is a big factor in determining valuable results. To greater or lesser extents, data of insufficient quality, polluted by meta noise and inconsistent formatting, could dramatically hinder the functionality of some models and lead to problematic analyses. So, in some cases more than others, data quality will be the principal consideration.
In a recent comparative study, a human ether-a-go-go related gene (hERG)-induced cardiotoxicity forecasting model was developed using 290,000 structurally diverse compounds collected from GOSTAR®, hERGCentral, and other open-source sources. The support vector machines (SVM) classification model of this study outperformed the available commercial hERG prediction software with an accuracy of 0.984 for the test set. These findings reinforce earlier research that has used larger data sets with diverse chemical space to develop predictive models with greater reliability and scope. [6-8]
The quantity and quality of available data, and the breadth of its chemical coverage, become essential when selecting a database to build and train AI/ML models on. Excelra’s GOSTAR® is the world’s largest database curated by domain experts, and its data quantity, quality, and coverage are exemplary. Comprehensively compiled and consistently structured, GOSTAR®’s data is also easy for a data scientist to prepare for a predictive model. In the next section, we outline a typical data set preparation process and underscore some important questions for a data scientist to consider.
Preparing GOSTAR® data for AI/ML models
Step 1: Select the data
This step involves selecting a subset of all available data. Though there may be an urge to include all available data, it isn’t always true that more data is better. More data is only necessarily better if it’s relevant to the study’s research question. Machine learning models can be misled if the training set is not representative of reality.
- Retrieve all data associated with the target(s) of interest:
- Define the target(s) using Common_Name, , UniProt ID, or similar fields available in GOSTAR®.
- Ensure that data obtained from searching target(s) of interest in step 1 have chemical structures associated with the results:
- Chemical structures are required since the primary focus is on the structure-activity relationship (SAR) and AI/ML models relating chemical structures to bioactivities.
- Rows of data retrieved which do not have associated chemical structures can be discarded for this exercise.
- Identify the experimental conditions:
- Ideally, data should be obtained from previous experiments conducted under similar experimental conditions to ensure a valid comparison.
- A conservative (and impractical) approach would be to only consider records that have identical experimental conditions. Including only these records would significantly reduce the data available for modeling.
Consider the following questions:
- How many of the experimental conditions’ fields available in GOSTAR® can be exact matches?
- Which conditions are similar enough to allow for combining data from multiple studies?
- How can we take advantage of studies which may not have explicitly recorded experimental conditions?
The answer to these questions is very subjective and requires agreement between data scientists and experimentalists. The experimental conditions in GOSTAR® reflect the information available in publications and patents. GOSTAR® enables rapid extraction of this information without having to read, analyze and curate data directly from hundreds or thousands of primary references.
Step 2: Pre-process and transform the data
The next step is to preprocess and transform the selected data. While the preprocessing step involves converting the selected data into a usable format, the transformation step is influenced by the algorithm used and understanding of the problem domain. Several iterative transformations to the processed data may be required, some of which are illustrated below.
Consideration of endpoint fields:
- Inspect search results to determine how the majority of data is recorded: IC50, EC50, %Inhibition, etc. Combining observations from different endpoints is not usually reasonable unless rules are defined for qualitative modeling. Identifying the most prevalent endpoint will increase the quantity of useful data.
- Activity prefixes must be considered and accounted for when combining results for modeling. Censured data, i.e., “<“, “>” must be removed or accounted for in continuous models. Categorical models are less sensitive than continuous models to censured data.
- measurement needs to be consistent and converted to “standard units,” which should be defined according to project needs. Only comparable units of measurement should be converted.
- Activity remarks should be considered, as this field highlights potential inconsistencies documented in the primary sources.
General Comments:
- The resulting data set must be carefully examined to remove rows that contain “null” or missing values if these values are critical to the modeling exercise.
- Structures must be standardized and prepared for descriptor generation. There are numerous approaches to chemical structure standardization in the literature, which cover topics such as salt stripping, tautomer generation, etc.
- Extreme outliers of both the experimental endpoints (outside typical ranges of measurement) and chemical structures (polymers, mixtures, etc.) should be removed.
- Quantitative models (regression) may not necessarily be required depending on the ultimate use case, e.g., classifying compounds into bins according to predetermined criteria.
- Data scientists should consider the chemical diversity of the data set and estimate the probability of new compounds falling inside or outside the chemical space from which the model was derived.
- Clearly define the intended purpose of the model before embarking on the derivation of a model. Data selection should be adapted to meet the requirements.
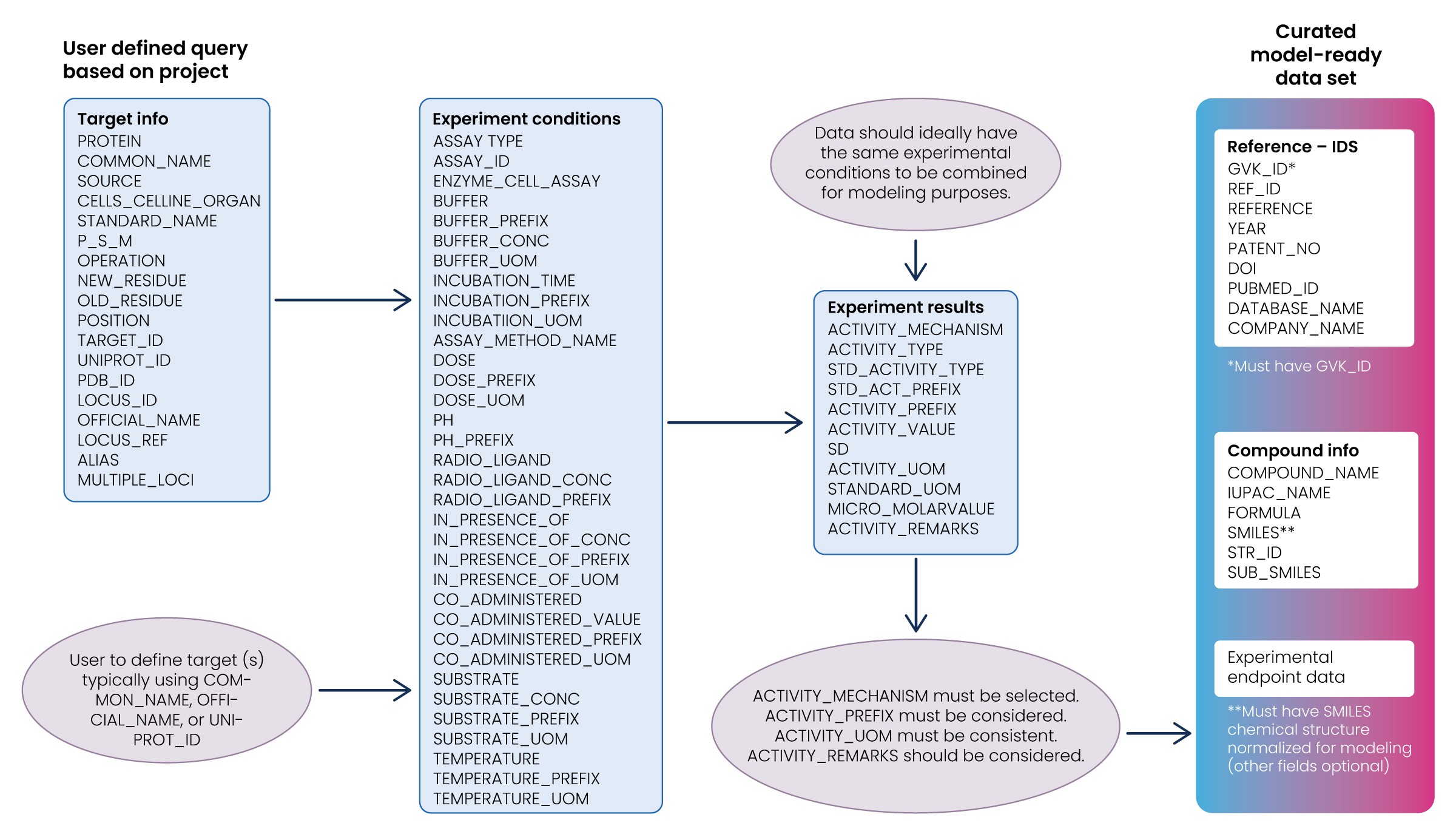
Figure 2. The process of curating GOSTAR® data for AI/ML applications
Conclusion
The democratization of artificial intelligence technology, combined with the introduction of big data and ever-increasing computing power, has accelerated AI/ML adoption, particularly in the pharmaceutical and biotech industries. However, the predictive accuracy of AI/ML models is dependent on the data that powers them. AI/ML advances are inhibited by data of insufficient quantity, quality, and coverage.
GOSTAR® overcomes this problem by delivering comprehensive, homogenized, high-quality data typically missing from other SAR databases. Users can take full advantage of the millions of compounds and associated endpoints in the GOSTAR® database by following a framework for selecting and preparing data. Once selected and prepared, GOSTAR® data can help build optimized predictive models that could uncover major discoveries.
GOSTAR® data alleviates bottlenecks, saves time, and improves accuracy. When technology depends on data, researchers depend on GOSTAR®.
To learn more about GOSTAR® and Excelra’s data, insight, and R&D technology services, visit excelra.com/GOSTAR®.
References
- Panteleev, J., Gao, H., & Jia, L. (2018, September). Recent applications of machine learning in medicinal chemistry. Bioorganic & Medicinal Chemistry Letters, 28(17), 2807–2815. https://doi.org/10.1016/j.bmcl.2018.06.046
- Vamathevan, J., Clark, D., Czodrowski, P., Dunham, I., Ferran, E., Lee, G., Li, B., Madabhushi, A., Shah, P., Spitzer, M., & Zhao, S. (2019, April 11). Applications of machine learning in drug discovery and development. Nature Reviews Drug Discovery, 18(6), 463–477. https://doi.org/10.1038/s41573-019-0024-5
- Dara, S., Dhamercherla, S., Jadav, S. S., Babu, C. M., & Ahsan, M. J. (2021, August 11). Machine Learning in Drug Discovery: A Review. Artificial Intelligence Review, 55(3), 1947–1999. https://doi.org/10.1007/s10462-021-10058-4
- Vijayan, R., Kihlberg, J., Cross, J. B., & Poongavanam, V. (2022, April). Enhancing preclinical drug discovery with artificial intelligence. Drug Discovery Today, 27(4), 967–984. https://doi.org/10.1016/j.drudis.2021.11.023
- Opidi, A. (2019, September 20). Solving Data Challenges In Machine Learning With Automated Tools. TOPBOTS. https://www.topbots.com/data-preparation-for-machine-learning/
- Sato, T., Yuki, H., Ogura, K., & Honma, T. (2018, July 6). Construction of an integrated database for hERG blocking small molecules. PLOS ONE, 13(7), e0199348. https://doi.org/10.1371/journal.pone.0199348
- Mayr, A., Klambauer, G., Unterthiner, T., Steijaert, M., Wegner, J. K., Ceulemans, H., Clevert, D. A., & Hochreiter, S. (2018). Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chemical Science, 9(24), 5441–5451. https://doi.org/10.1039/c8sc00148k
- Plonka, W., Stork, C., Šícho, M., & Kirchmair, J. (2021, September). CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes. Bioorganic & Medicinal Chemistry, 46, 116388. https://doi.org/10.1016/j.bmc.2021.116388
How can we help you?
We speak life science data and help you unlock its potential.