“Effective target identification lies at the core of drug discovery. In theory, it sounds simple — find a target, match it with a game-changing therapy. In practice, confirming that match is a scientific quest in itself”
Introduction
Determining the ability of a target to bind to a drug with high affinity to modulate its function with a therapeutic benefit could be termed as Druggability. It needs a non-trivial amount of time, financial investment, and resources. An investment of US$2.6 billion (£1.9 billion) over 10-15 years for inventing a new drug from target identification to approval, which ultimately benefits the patients, underscores the importance of prioritizing the most potential druggable targets.
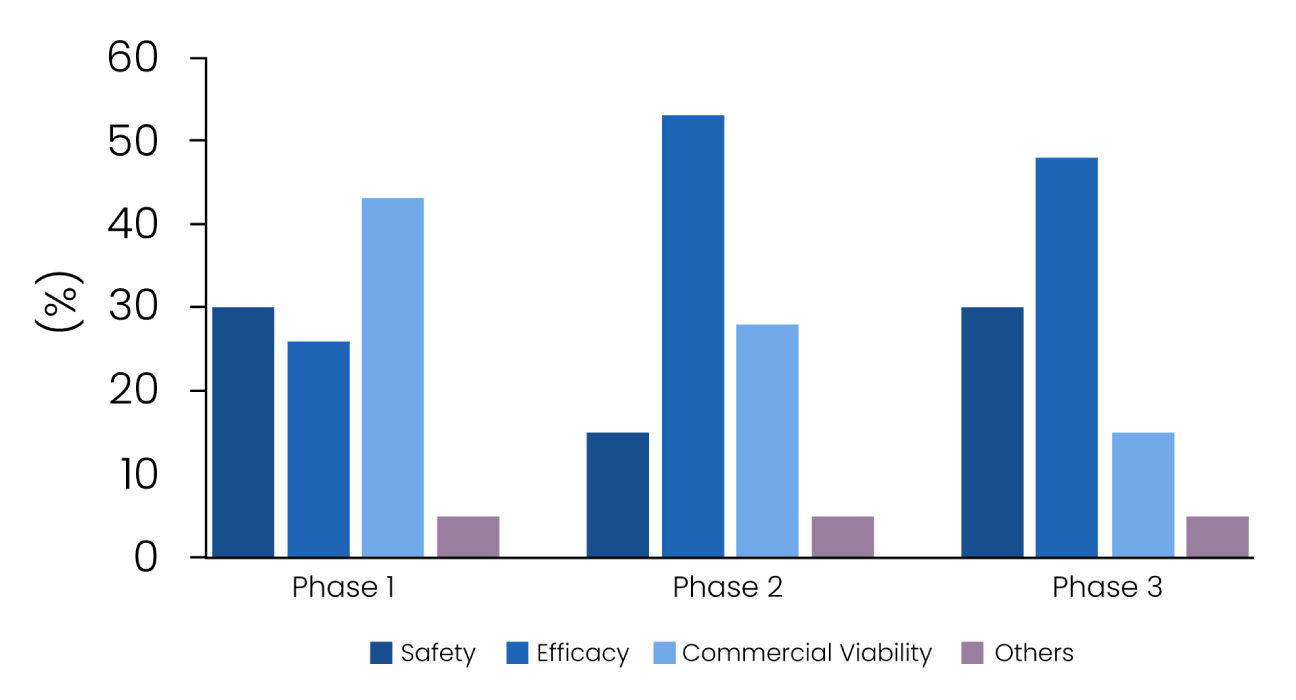
Figure 1: Reason of clinical trial failures by phase
Comparable to the high cost, the success rate for drug development programs is substantially low with over 90% of the drug candidates fail in clinical trials, which put the pharmaceutical industry under mounting pressure. The biggest reason for drug attrition is mainly safety findings or lack of efficacy (Figure 1), nearly 60% of failures are attributed to inappropriate target identification and validation in drug discovery.2,3 To increase the chances of success and maximize return on investment, drug inventors must consider strategies focusing on identification of the most biologically plausible druggable targets that effectively modulate the disease phenotype on a molecular level.
“The most potential targets must have strong association with the disease of interest, an established role in the mechanism of disease pathology, druggability or chemical tractability, and biological tractability and assayability”
Selecting the right target early in the discovery R&D process is crucial in enabling the prospects of regulatory approval success. Prioritization of most promising targets involves consideration of several key factors including disease relevance, role in underlying pathophysiology, specificity to the disease process or state, high frequency in the patient population, tractability (the possibility of finding small molecule compounds with high affinity), safety profile, novelty, as well as the competitiveness in the market (Figure 2). Subsequent to target identification, the critical imperative is to assess target druggability which is integral to successful target validation and is a pivotal step in every drug design quest. Despite the known 3D protein structures surpassing 100,000 in number and ChEMBL database encompasses around 5,000 known proteins with bindable pockets, a mere 854 of these proteins have been recognized as established therapeutic targets for FDA approved drugs. These data evidently demonstrate that the eligibility of a protein, and consequently the protein pocket, for binding of a molecule and hence drug discovery is a formidable challenge. Additionally, because a substantial number of small-molecule drug discovery project failures are owing to poor bioavailability, careful evaluation of biological and molecular basis of target druggability and building a precise druggability prediction method (DPM) are extremely essential at the early stage of drug discovery programs.
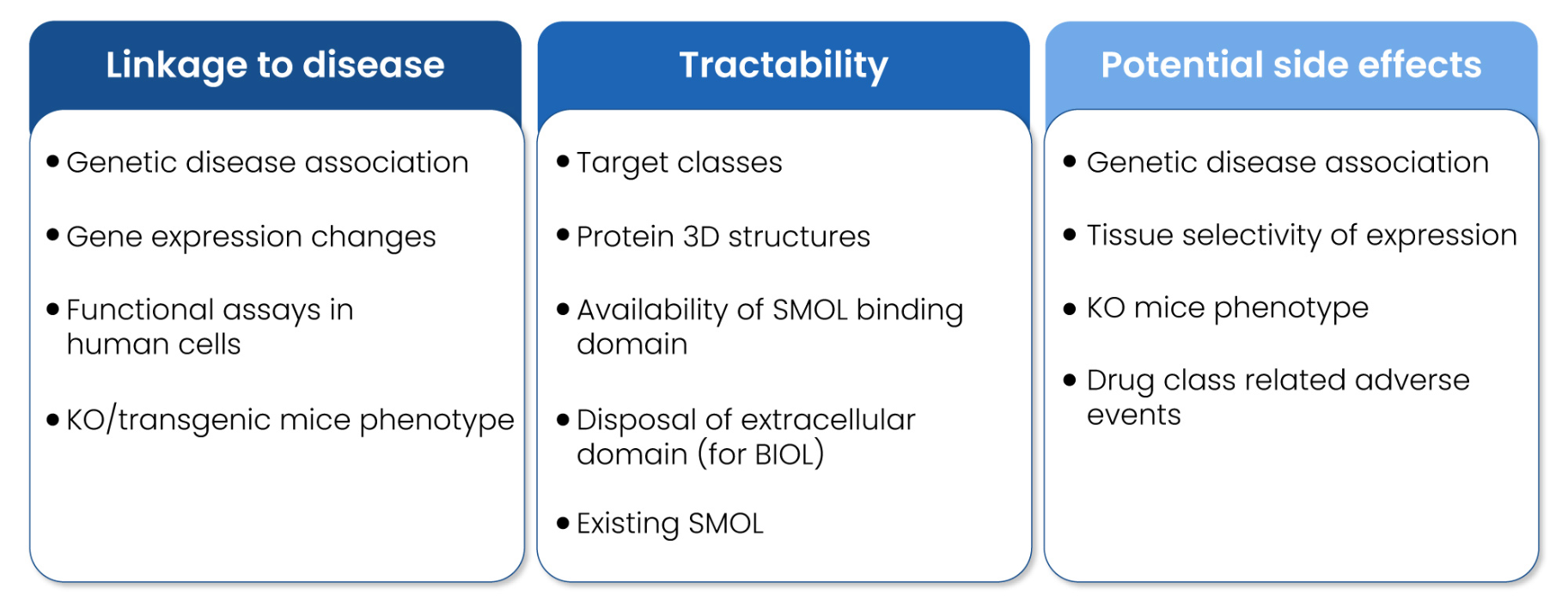
Figure 2: Key factors to be considered in target prioritization
“Targeting the undruggable proteome – Targeted Protein Degradation – a therapeutic strategy that co-opts the cell’s degradation machinery (UPS or Autophagy) to target disease-causing proteins”
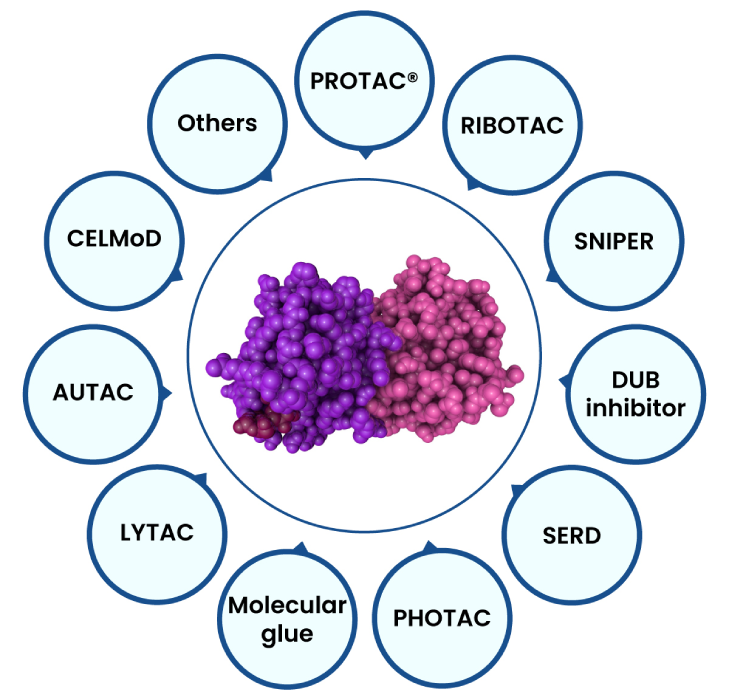
Figure 3: Different degrader class
The complexity mounts as there are approximately 20000 human protein-coding genes, but not all proteins are suitable for drug interactions and even fewer are appropriate drug targets, because the lack of so-called gene knockout methods and druggable deep grooves and active pockets to serve as the binding site for small molecules, such as scaffolding proteins, transcriptional factors, and RAt Sarcoma (RAS) proteins were deemed “undruggable” proteins in the past. To address these challenges several big pharma companies (such as Novartis, Pfizer, AbbVie, Bayer) and PROTAC® companies gained foothold in a new technology called targeted protein degradation (e.g., PROTAC®, Molecular Glue, etc) (Figure 3), which offers a unique means to tap into the rest of the vast, unexplored proteome. A PROTAC® can bind to target proteins without the presence of active pockets, leading to proteasome mediated degradation and complete inhibition of the biological functions of the target.
This concept is therefore an attractive investment opportunity harbouring the potential for blockbuster medications within therapeutic domains including cancer and neurobiology, thereby addressing targets previously deemed ‘undruggable.’
Reducing Drug Attrition Rate
Selection of right target
Selecting an appropriate target marks the initial and indispensable step to launch a discovery program. Likelihood of an experimental drug to become a clinical candidate or successfully entering the market is exceedingly low with attrition rate escalating every year. Among the primary contributors of the drug failure are inappropriate target selection, insufficient elucidation of mechanism of action, safety concerns, and sub-optimal efficacy. Implementing effective strategies for therapeutic target identification can significantly reduce the time and efforts that drug hunters invest in the crucial stage. Target identification strategies (Figure 4) include genetic associations (connecting genes with a disease), proteomics, (activity-based protein profiling), data mining methods for searching through literature and databases. Other key factors contributing to successful target identification are understanding of disease biology, their molecular mechanisms and accessibility of predictive models and upholding technologies and assessment of target-based toxicity.
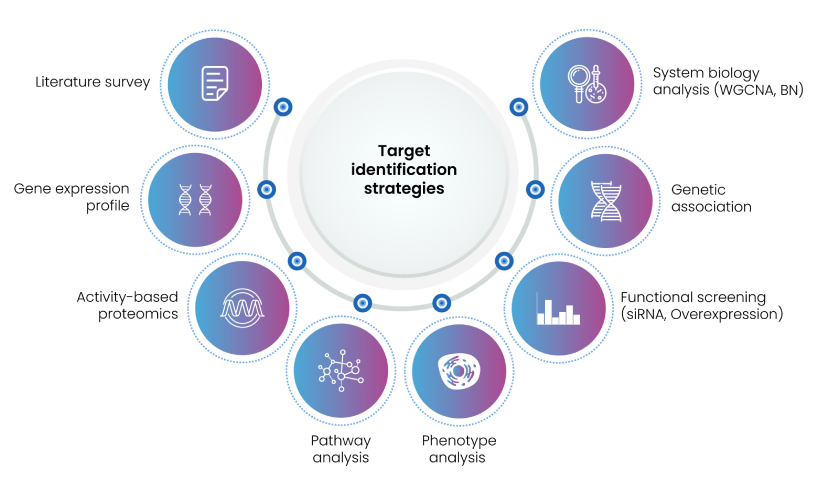
Figure 4: Target identification approaches
Target related safety liabilities
A rigorous assessment of the potential hazards induced by target modulation is a crucial part of a successful drug discovery program. The goal of a target safety assessment is to identify potential unintended adverse consequences of target modulation, and to propose a risk evaluation and mitigation strategy to guide molecules through the discovery and development pipeline. Deep understanding on the safety profile of the targets in the discovery pipeline is essential to make informed decisions that maximize the chances of success. Adverse events of kinase inhibition have ranged from the manageable (e.g., skin rash arising from EGFR inhibition) to the potentially severe or life threatening (hypertension following VEGFR inhibition). Similarly, 5-HT2B receptor agonism is known to show off-target risk of cardiac valvulopathy.4-6 The target safety assessment entails a comprehensive review of the target encompassing various aspects (Figure 5) of the biology (gene, protein, function, pathway, expression profile, tissue distribution, disease, etc), human genetic phenotype, transgenic animal genotype and phenotype (knock-out [KO], mutants), potential safety risks and its associated mitigation plans, and strategic elements like competitive intelligence and differentiation criteria.
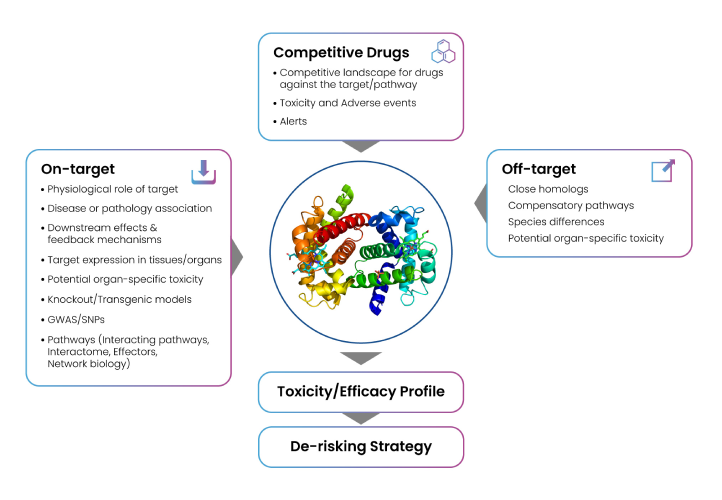
Figure 5: Workflow of target safety assessment
Druggability of the target of interest
Precise predictions of protein druggability and the impact of gene knockout play a pivotal role for selection of drug targets in early stages of drug discovery.
Experimental druggability assessments involved high-throughput or NMR-based screening using libraries of molecules that adhere to lead-like characteristics. Nonetheless, expensive instrumentation and the ambiguity stemming from negative assessments due to potentially inadequate compound libraries limit the scope of experimental approaches. Therefore, numerous computational procedures, typically based on virtual screening and machine learning, have emerged in the recent decades. Four primary methods are frequently employed for assessing druggability: which includes sequence-based, structure-based, ligand-based, and precedence-based methods to assess druggability (Figure 6).
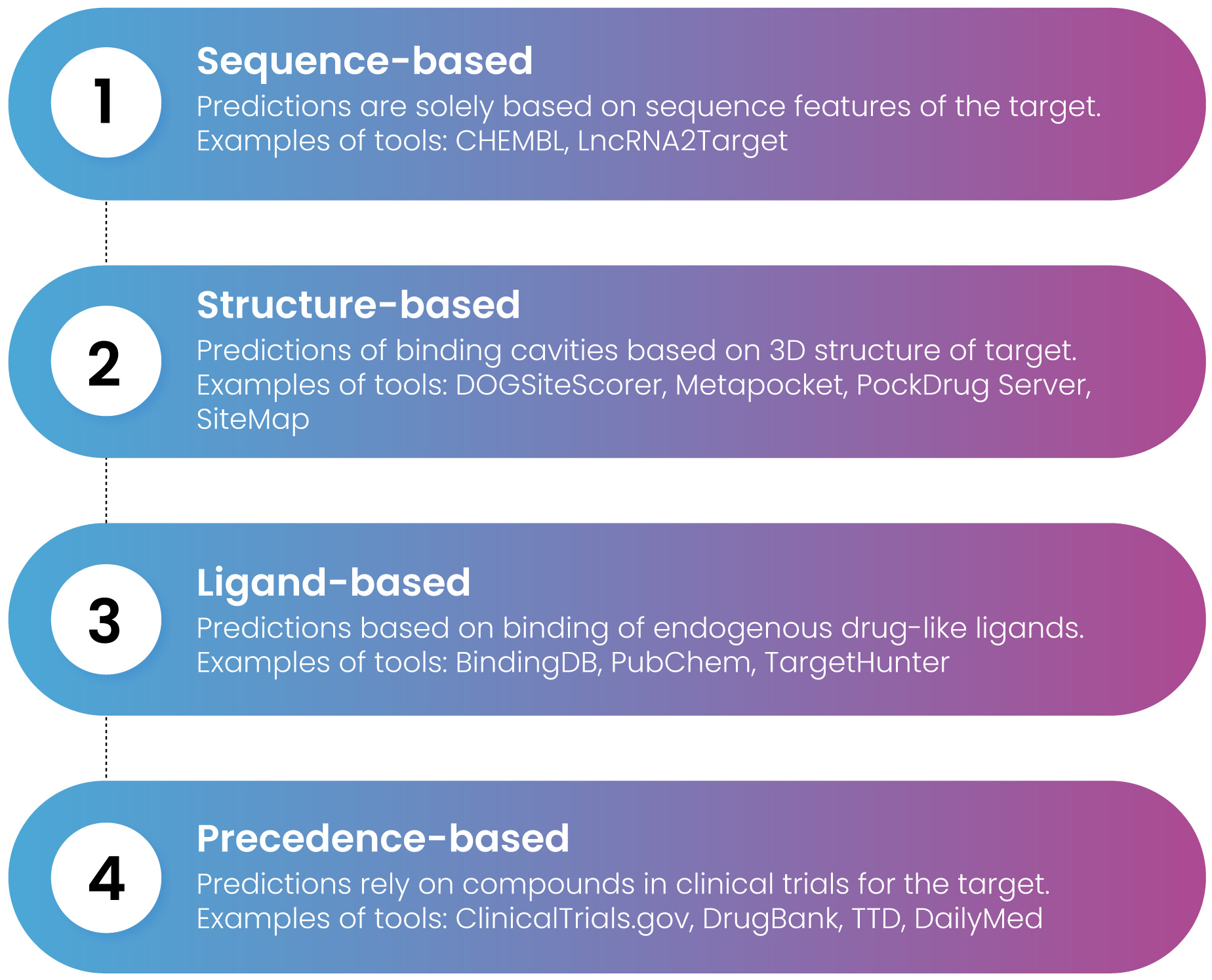
Figure 6: Methods of assessing druggability
Virtual screening is a knowledge-driven, compound database searching approach that uses computational approach to identify new lead compounds and chemotypes with a required biological activity. This approach is broadly categorized into two types, namely, structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS). Numerous studies offer innovative usage of virtual screening to specific biological targets, frequently in combination with other techniques. Pérez-Sánchez and co-workers depicted DIA-DB,7 a web server for the prediction of discovering diabetes drugs through ligand-based similarity or inverse docking against a set of relevant protein targets.7 Liang and co-workers showcased the discovery of covalent binders to the ATP binding pocket of PLK1 through virtual screening of a natural product database.8 Li and co-workers presented a computational approach combining, key interaction pattern recognition, virtual screening, and fragment-based design to the discovery of novel ligands against two GPCR targets β2-AR and mAChR M1.9 While the technique has gained widespread adoption, there remain significant areas for improvement. A notable limitation resides in the scoring functions used in docking. To evade the challenge, new machine-learning-based scoring functions have also been developed for docking as alternative to the classical scoring functions. In addition, the traditional in silico approaches are hampered by several weaknesses including reduced accuracy, high costs and time requirements, lack of data integration, human bias, and inability to handle large datasets. These shortcomings propel the shift in paradigm of druggability assessment from conventional in silico to machine-learning-based approaches.
Machine-learning (ML) based methodologies can handle vast datasets, extract complex patterns, provide more accurate predictions, and reduce human bias, ultimately speeding up the druggability assessment. Various machine learning algorithms have been employed to develop in silico models for the druggability assessment (Figure 7), which help to analyse biological data, predict molecular glue interactions, and identify potential drug candidates.10
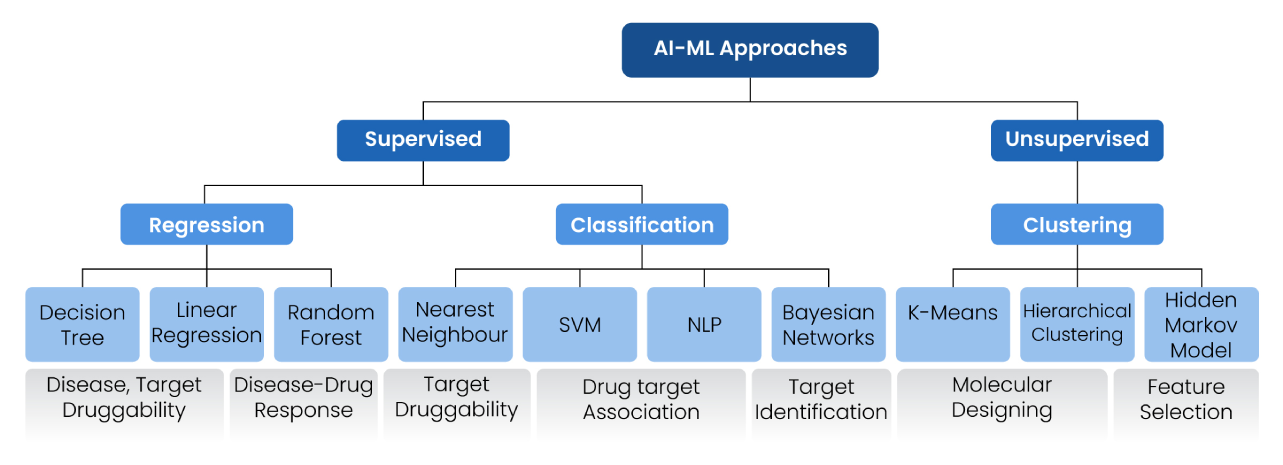
Figure 7: Methods to predict the druggability based on machine learning
Random forests, support vector machines, and gradient boosting are common machine learning models that are used to predict druggability based on features like protein structure, sequence, high throughput virtual screening and interaction data. AI based tools like Alphafold also help in protein structure prediction, ligands binding affinity and protein-ligand interactions quantification. Deep neural networks and convolutional neural networks (CNNs) are used to identify protein-protein interactions, protein-ligand binding, and drug-target interactions. Fingerprint analysis can also be performed using NLP. Deep generative models are used to generate novel molecular structures that have druggable properties. They enable researchers to explore a wider range of potential drug targets, locate groups of residue organization that build an active site, predict interactions, and prioritize candidates for further experimental validation, ultimately speeding up the drug discovery process. AI-ML based methods can also be used to analyse protein-protein interaction networks, genomic and transcriptomic data to identify genes and pathways associated with diseases. To address the druggability of the flexible proteins, protein modelling in concert with druggability estimation along with light protein backbone movement and side-chain flexibility in target binding sites are considered. To achieve better druggability scores, rigid-body movements in pockets coupled with structure based druggability analysis are commonly performed in most AI based approaches. In one success story, Yang et al. constructed a protein-protein interaction network for thyroid cancer and identified three key targets, HEY2, TNIK, and LRP4. They further used PockDrug, which is a novel web server that is employed to predict pocket druggability on proteins, to predict whether HEY2, TNIK, or LRP4 have targetable pockets. Based on the predictions, TNIK, which has 8 out of 538 residues, has an average probable score of druggability greater than 0.5 and thus was considered a druggable pocket for thyroid cancer.11 Taken together, the advancement in Machine-learning algorithms can quickly and accurately determine the druggability of targets, provide reliable druggable targets for indication of interest, and reducing the time and financial costs of experiments.
Unmet need & commercial essence of a target of interest
Before committing to new drug development for certain disease, pharmaceutical and bioinformatics services companies investigate the drug landscape for the target. After analysing various facets of competitive intelligence, the companies can make an informed go/no-go decision.
Pharma companies ideally survey the target landscape to identify market opportunities, review previous failed attempts, and collect all the knowledge. Increasingly, they seek to understand target-compound landscape to gain comprehensive, verified, high-quality insights to support the decisions they will make as their research program progresses. Drug landscape assessment for the target gives understanding about preclinical, clinical, and approved drugs and their mechanism of action towards the target (Figure 8). The assessment also provides a holistic view of ongoing global research on target of interest and different diseases wherein target plays a key role. Understanding the preclinical space allows the companies to peruse through voluminous data from patents and publications to understand chemical moieties to circumvent issues related to intellectual property etc.
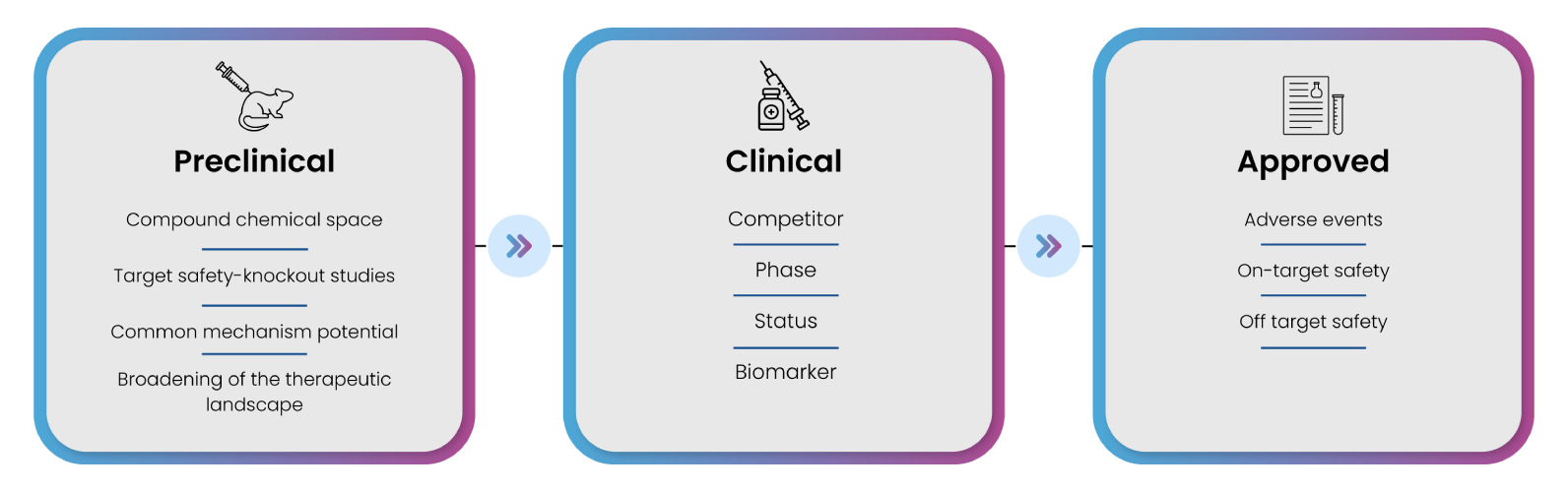
Figure 8: Vital information that can be retrieved from the different stages of drug development
It also helps them to design novel chemical entities rationally. Clinical Trials Intelligence provides granular information so that company allows its team to quickly identify competitor trials related to compounds/different modalities aiming at the target of interest. The analytics and alerts allow them to visualize a competitor’s design and timeline. This gives them confidence in their standing relative to competitors and helps to guide clinical trial strategy/decisions. To succeed in the pharmaceutical marketplace, it is vital to stay up to date with strategies/developments related to a particular target. For instance, target-compound landscape assessment potentially reveals new strategies of targeting a protein of interest. It may open new modalities such as allosteric binders, and small molecules inhibitors which disrupt protein-protein interactions, degraders etc. thereby providing novel therapeutic strategies for suppressing the target’s functional role in pathogenesis. The assessment also confirms whether the target is having first-in-class compounds, informing ongoing commercial and intellectual property (IP) strategies.
Overall, Drug landscape assessment can play a great role in the investment decision of whether to pursue a project. Increasing awareness of target-compound landscape and competitor activities help to determine how to differentiate the drug candidate in the market to be successful. This assessment captures all relevant information covering the successes and failures of targeting the protein of interest, drugs in preclinical and clinical stages, competing drugs in the market, and salient features of ongoing clinical trials.
Transforming undruggable proteins into therapeutic targets
A vast majority of the human genome remains undruggable, amounting to roughly 85-90% of all human proteins. FDA approved drugs target only a little over 800 proteins out of the roughly 20 thousand odd proteins in the humans (Figure 9) and around 2 to 3 thousand have a small molecule or a biologic under clinical development targeting them.
Drug discovery and development is still profoundly dependent on the use of molecules capable of occupying a specific active site on the target protein, with direct consequences on its function. Therefore, it is difficult to develop a proper strategy in the absence of such defined pockets. Other characteristics of undruggable proteins include, highly disordered regions, lack of possibility of catalytic or competitive inhibition, large protein-protein interaction (PPI) interfaces etc (Figure 9).
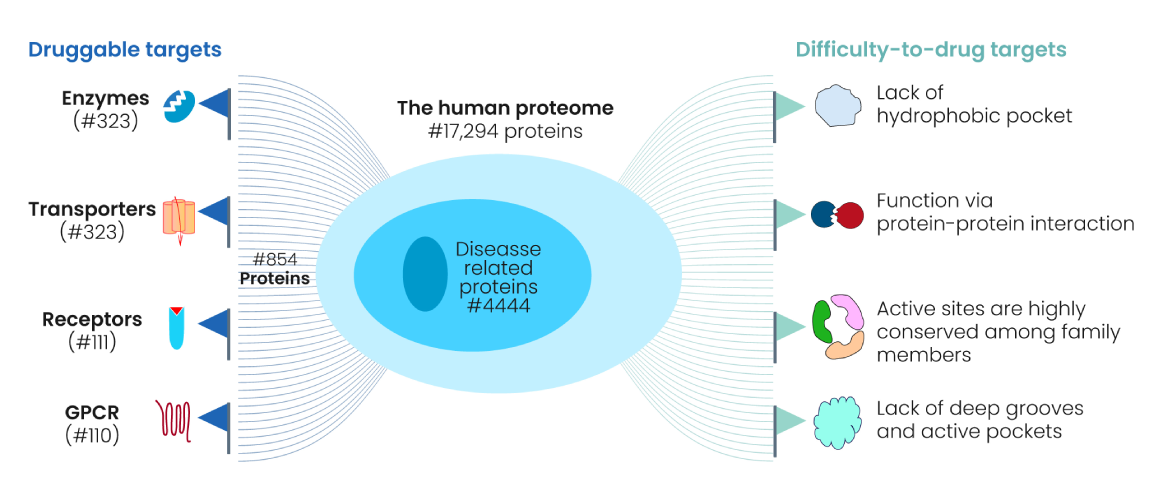
Figure 9: Druggable and undruggable human proteome
At Excelra, we are at the forefront of analysing new methodologies for evaluating druggability of targets and targeting the undruggable proteins. One of the recent developments is the advent of Targeted Protein Degradation methodologies. The archetypal type of molecules are PROTAC®’s which are hetero-bifunctional small molecules. They bind simultaneously to a target protein and an E3 ubiquitin ligase, thereby causing ubiquitylation and degradation of the target protein via the 26S proteasome (Figure 10).
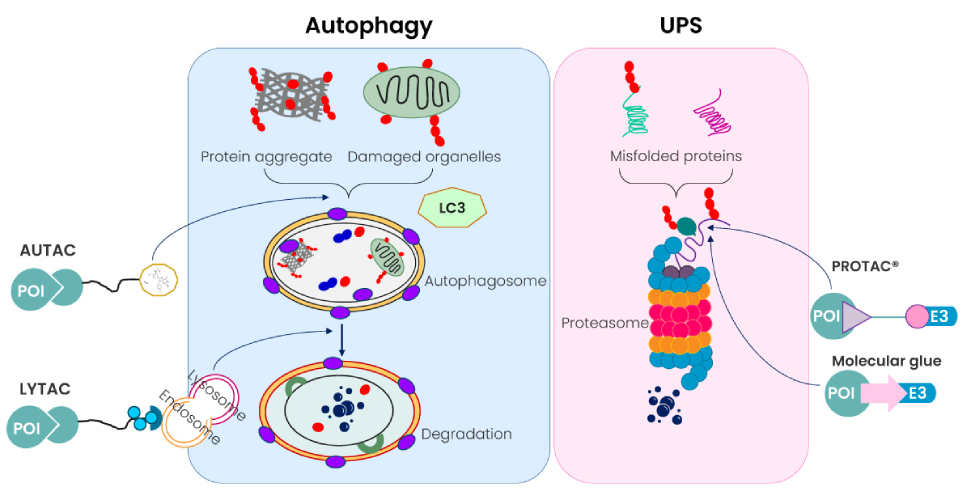
Figure 10: Cellular protein degradation pathways and novel targeted protein degradation modalities
The ability of PROTAC®’s to target and degrade proteins, regardless of their function, makes this approach highly beneficial, especially for those targets for which compounds can be developed that bind to a given target without inhibiting its activity. Degradation of the target protein by PROTAC®’s is also suitable for those proteins that overcome the effect of an inhibitor by overexpression, which is often observed in cancer.
PROTAC®’s have several advantages; because of their unique mechanism of action, a PROTAC® molecule is capable of degrading multiple molecules of the protein of interest. Also, in comparison to small-molecule inhibitors, PROTAC®’s have been shown to require significantly lower concentrations to elicit a desired pharmacologic effect. PROTAC®’s also have the potential to target so-called undruggable proteins, such as transcription factors (For e.g. STAT, Myc) and oncogenes (For e.g. KRAS). PROTAC®’s can be used to overcome drug resistance resulting from mutations of a protein of interest (For e.g. Bcr-Abl).
Figure 11: Challenges in TPD space
As per recent count, 18 PROTAC®’s are under clinical development with ARV-471 being investigated in Phase-3 clinical trials. More importantly, Kymera Therapeutics’ STAT3 PROTAC® KT-333 received fast track designation from the FDA to treat Relapsed/Refractory (R/R) Cutaneous T-Cell Lymphoma (CTCL) and Peripheral T-Cell Lymphoma (PTCL).
Although PROTAC® technology has a bright future in drug development, but it also has many challenges (Figure 11). Excelra has created a Degrader Database that covers Chemistry datapoints, Biological endpoints, and Competitive intelligence that can be leveraged to kickstart a new program or fine tune the existing assets. The data are captured from several authentic sources, including scientific journals, patents, company pipelines, conference presentation, databases, etc.
Conclusion
A drug discovery program starts with identification of novel and effective drug development technology with biological targets for the development of new drugs with the unmet clinical need. Discovering and evaluating the potential therapeutic benefit of a drug target is founded not only on experimental, mechanistic, and pharmacological studies but also on a theoretical molecular druggability assessment, an early evaluation of potential safety measures and through considerations regarding opportunities for commercialization as well as options for generation of IP. Traditional approaches are inadequate for large-scale exploration of novel drug targets, as they are expensive, time-consuming, and laborious. In recent years, various computational strategies for predicting potential druggable proteins have emerged, which commonly use the sequence, structural, and functional features of proteins as input but also system-level properties such as network topological features. Despite tremendous success, unfortunately many promising and experimentally validated targets are not within the scope of drug modifiability. Discovery of next generation technologies including targeting protein degradation, protein stabilizers (RESTORACs), excellent drug delivery system, targeting PPI, targeting intrinsically disordered regions, as well as targeting protein-DNA binding may provide significant assistance in overcoming these undruggable targets.
References
1. DiMasi, J. A., Grabowski, H. G. & Hansen, R. W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20-33 (2016)
2. Laenen, G., Thorrez, L., B¨ornigen, D., & Moreau, Y. Finding the targets of a drug by integration of gene expression data with a protein interaction network. Molecular BioSystems. 9(7), 1676-1685 (2013)
3. Arrowsmith, J. Phase III and submission failures: 2007-2010. Nature Reviews Drug Discovery. 10(2), 1-2 (2011)
4. Liu S, Kurzrock R. Toxicity of targeted therapy: implications for response and impact of genetic polymorphisms. Cancer Treat Rev. 40(7), 883–891 (2014)
5. Liu S, Kurzrock R. Understanding toxicities of targeted agents: implications for antitumor activity and management. Semi n Oncol. 42(6), 863–875 (2015)
6. Small HY, Montezano AC, Rios FJ, Savoia C, Touyz RM. Hypertension due to antiangiogenic cancer therapy with vascular endothelial growth factor inhibitors: understanding and managing a new syndrome. Can J Cardiol. 30(5), 534–543- (2014)
7. Perez-Sanchez H, den-Haan H, Peña-García J, LozanoSanchez J, et al. DIA-DB: A database and web Server for the prediction of diabetes drugs. J. Chem Inf Model. 60(9), 4124-4130 (2020)
8. Liang H, Liu H, Kuang Y, Chen L, Ye M, Lai L. Discovery of targeted covalent natural products against PLK1 by herb-based screening. J. Chem Inf Model. 60(9), 4350-4358 (2020)
9. Li Y, Sun Y, Song Y, Dai D, Zhao Z, Zhang Q, Zhong W, et al. Fragment-Based Computational Method for Designing GPCR Ligands. J. Chem Inf Model. 60(9), 4339-4349 (2020)
10. Abi Hussein H, Geneix C, Petitjean M, Borrel A, Flatters D, Camproux AC. Global vision of druggability issues: applications and perspectives. Drug Discov Today. 22, 404–415 (2017)
11. Yang YF, Yu B, Zhang XX, Zhu YH. Identification of TNIK as a novel potential drug target in thyroid cancer based on protein druggability prediction. Medicine. 100:e25541 (2021)
How can we help you?
We speak life science data and help you unlock its potential.