
Matched molecular pair analysis addresses the complexity in molecular design when selecting what to do next based on existing data, medicinal chemistry knowledge, experience, and intuition. In small compound sets, a skilled chemist can discern trends and correlations by eye. As the number of molecules increases, more methodical procedures are required.
Matched molecular pair analysis
The Matched Molecular Pair (MMP) analysis, which compares closely related chemical structures pairwise across a big dataset, is one method in the medicinal chemist’s toolbox for accomplishing this. Since the structures of the two molecules in question differ very slightly, any change in a physical or biological feature between the matched molecular pair can be more easily interpreted.
In 2004, Kenny and Sadowski coined the term Molecular Matched Pair (MMP) for a subset of QSAR; it is now a widely used concept in drug design processes [1]. Matched molecular pairs differ only in small single-point alterations, which are referred to as chemical transformations. As the structural difference between the two molecules is minimal, any differences in physical properties or observed biological effects can simply be linked to it.
In 2010, Hussain and Rea published an approach to find matched molecular pairs and relate them to the distribution of value differences for each transformation, and it has since become a popular tool for analyzing huge chemistry datasets using matched molecular pair analysis.
Role of MMP in medicinal chemistry
MMP is typically used to describe a pair of compounds that differ structurally at a single site because of a well-defined transformation accompanied by a change in a property value. To rationalize observed structure-property relationships (SPR) and compound optimization, the relationship between structural and property change is used.
Aside from assisting in hypothesis creation and testing, matched molecular pair analysis can also be used to find outliers, such as a pair of compounds that have a sudden change in a property, known as an activity cliff. These compounds are typically the most intriguing to investigate in the development of compounds aimed at increasing the property that exhibits this change.
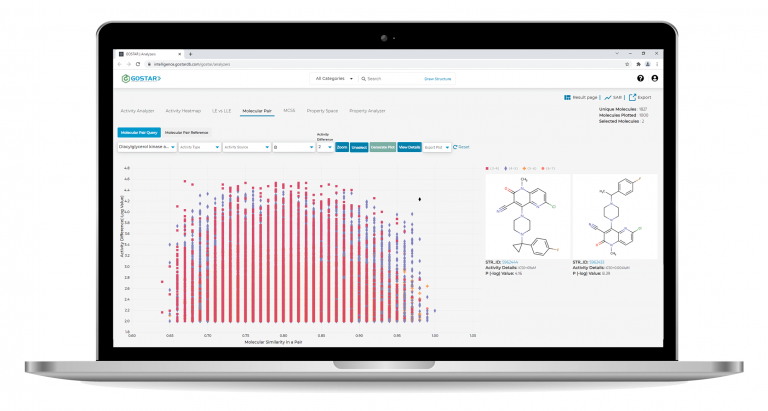
Matched molecular pair analysis in GOSTAR™
GOSTAR™ provides tools for determining the matched molecular pairs and analyzing activity landscapes across compound datasets. The matched molecular pair analysis capability within GOSTAR™ supports medicinal chemists in understanding structure–property trends, identifying activity cliffs, and enabling data-driven compound optimization.
This analytical functionality is part of Excelra’s broader GOSTAR™ platform, which integrates medicinal chemistry, SAR, and biological activity data to accelerate data-driven drug discovery and advanced analytics in medicinal chemistry.
References
1. Kenny P.W., Sadowski J. Structure modification in chemical databases. In: Oprea T., editor. Cheminformatics in drug discovery. Wiley-VCH Weinheim; Germany, 2004, 271.
2.Hussain J, Rea C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J Chem Inf Model. 2010, 50(3), 339-348.
If you are new to GOSTAR™ and want to power your drug discovery with gold-standard data, request a demo now.
