
Authors: Sirisha Addanki (Associate Director, Sales) & Hariprasad Reddy Gadi (Director, ClinPharm)
In drug development, a clinical trial’s value does not end with the last patient visit. It continues for years—every time a regulatory reviewer, HTA analyst, or evidence synthesis team needs to extract structured clinical, safety, and efficacy data from published literature to feed into systematic reviews, meta-analyses, or regulatory dossiers. This process is foundational to scientific data management and modern scientific informatics services for life sciences.
This extraction process—spanning clinical outcomes, safety signals, efficacy endpoints, and regulatory evidence—is one of the most consequential and least automated steps in pharmaceutical evidence generation. It sits upstream of every payer submission, every Meta-Analysis model, and every competitive intelligence deliverable. And for most organizations in 2026, it is still done manually, limiting data readiness for drug discovery and downstream analytics. In the evidence generation process, data extraction is the most critical and time-consuming step.
The source material
A single completed Phase 3 trial typically generates four distinct source types: the primary publication; a supplementary appendix often as long as the main paper; a ClinicalTrials.gov posting that frequently contains data not published elsewhere; and conference abstracts (EULAR, ACR, ASCO, ASH) reporting interim or post-hoc results.
For a single indication with 8–15 relevant trials, an SLR/TLR team may work across 30–60 source documents simultaneously, contributing to complex data landscape assessment in pharma and broader disease landscapes analysis.
The current extraction process
The pipeline that medical writers and scientific SMEs execute today when preparing a structured dataset for meta-analysis or HTA use involves six stages and typically takes four to six weeks per indication.
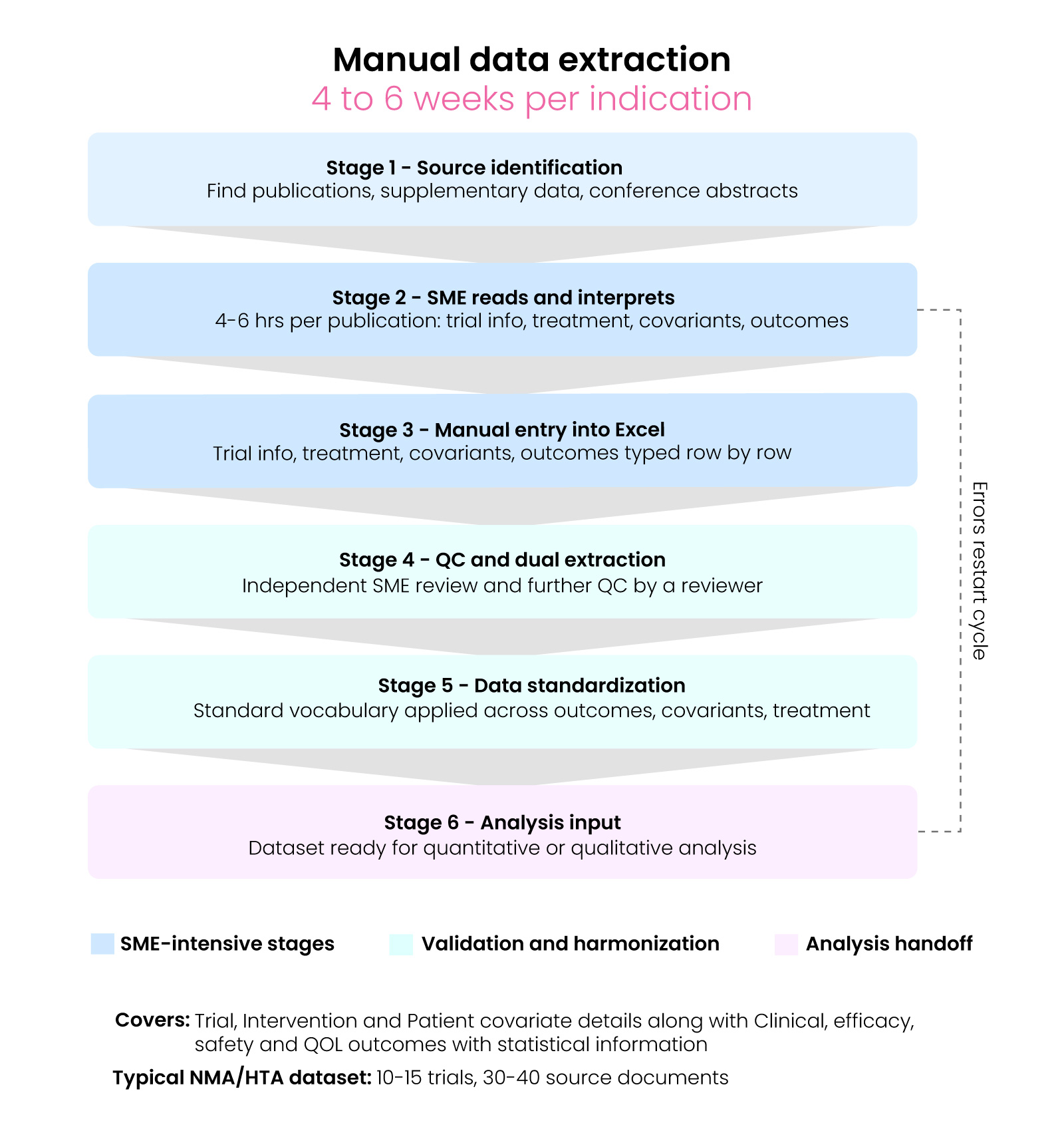
Stage 1 — Source identification
After screening, each included study requires finding the relevant publications — main paper, supplementary data, CT.gov record, and conference abstracts — located and organized across inconsistent databases.
Stage 2 — SME reads and interprets
A trained SME identifies the reporting context for every data point, capturing trial-level information, treatment details, patient covariants, and outcomes — including population, imputation method, and whether the value is the primary analysis or a sensitivity run. This takes 4–6 hours per publication.
Stage 3 — Manual entry into Excel
The SME types each parameter — trial-level information, treatment details, patient covariants, outcomes (point estimate, CI bounds, p-value, sample size, population definition, imputation code, time point, treatment arm) — one row at a time, with no software link to the source text.
Stage 4 — QC and dual extraction
Standard SLR/TLR methodology recommends dual independent. The standard SLR/TLR methodology recommends dual independent extractions. SMEs review the data, and further QC is performed by a reviewer, adding a second full extraction cycle per paper plus resolution time.
Stage 5 — Data standardization
A schema is put in place, and a standard vocabulary is applied across patient outcome details, covariant details, and treatment details. Treatment arm names written inconsistently across publications are mapped to canonical labels, and population definitions and imputation codes are cross-checked. For a 12-trial dataset, this step alone takes several days. This aligns with cro data standardisation and fair data principles in life sciences to ensure consistency and interoperability.
Stage 6 — Analysis input
The final dataset is ready for the quantitative or qualitative scientist. Any errors or provenance gaps discovered here require the SME to return to source documents, restarting the cycle.
Where it breaks down
The first failure mode is missing provenance. Every extracted value in a regulatory or HTA submission must be traceable to a specific table, footnote, or statistical method in the source document. Without that link documented at the point of extraction, a single question from a reviewer or data modeler triggers a full re-read of the original paper- under submission deadline pressure, for every challenged data point.
The second is schema drift. In a Meta-Analysis, every treatment arm must mean the same thing across all studies. When two SMEs read the same dosing description differently, the model ends up comparing interventions that are not actually equivalent- silently, with no error flagged, and no warning in the submission.
Why existing tools have not solved this
Established platforms such as Covidence, DistillerSR, and EPPI-Reviewer digitize screening workflows but do not perform extraction. The bottleneck—manual SME effort—remains unchanged.
This gap has driven the rise of AI agents in life sciences and AI agents for scientific informatics, enabling automation in data extraction, structuring, and validation.
Early LLM-assisted extraction experiments show promise, especially when combined with hybrid RAG for clinical programming and structured validation layers.
The direction of travel
A credible AI-integrated data curation implementation requires:
A credible AI-integrated data curation implementation requires: full provenance linking every extracted value to its source text; schema enforcement producing Meta-Analysis and Regulatory submission-ready
Trial, Intervention and Patient covariate details along with Clinical, efficacy, safety and QOL outcomes with statistical information. The question is no longer whether AI-assisted extraction will become the standard- it is which implementations meet the evidence-quality bar that pharmaceutical companies, regulators, and HTA bodies rightly demand.
This is where bioinformatics solutions and scientific informatics solutions converge with automation, enabling scalable therapeutic workflow digitalization.
The future lies in combining cloud solutions for scientific informatics, cloud-native lab informatics, and AI-driven pipelines to transform evidence generation at scale.
References
- U.S. National Library of Medicine. ClinicalTrials.gov — FDAAA 801 and the Final Rule.
- Higgins JPT, Thomas J, et al. Cochrane Handbook for Systematic Reviews of Interventions, Version 6.4. Cochrane, 2023.
- Page MJ, McKenzie JE, et al. The PRISMA 2020 statement. BMJ. 2021;372:n71.
- Fraenkel L, et al. 2021 ACR Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care & Research. 2021;73(7):924–939.
- Agrawal M, et al. Large language models are few-shot clinical information extractors. EMNLP 2022.
